
Sickle cell anemia
Overview
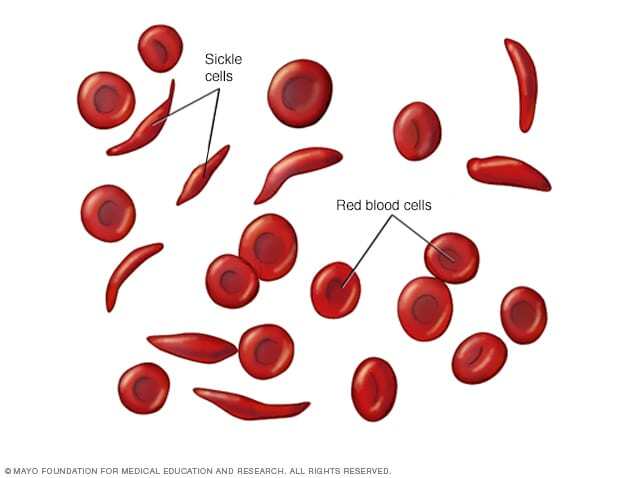
Sickle cell anemia is one of a group of inherited disorders known as sickle cell disease. It affects the shape of red blood cells, which carry oxygen to all parts of the body.
Red blood cells are usually round and flexible, so they move easily through blood vessels. In sickle cell anemia, some red blood cells are shaped like sickles or crescent moons. These sickle cells also become rigid and sticky, which can slow or block blood flow.
The current approach to treatment is to relieve pain and help prevent complications of the disease. However, newer treatments may cure people of the disease.
Symptoms
Symptoms of sickle cell anemia usually appear around 6 months of age. They vary from person to person and may change over time. Symptoms can include:
- Anemia. Sickle cells break apart easily and die. Typical red blood cells usually live for about 120 days before they need to be replaced. But sickle cells usually die in 10 to 20 days, leaving a shortage of red blood cells. This is known as anemia. Without enough red blood cells, the body can't get enough oxygen. This causes fatigue.
-
Episodes of pain. Periodic episodes of extreme pain, called pain crises, are a major symptom of sickle cell anemia. Pain develops when sickle-shaped red blood cells block blood flow through tiny blood vessels to the chest, abdomen and joints.
The pain varies in intensity and can last for a few hours to a few days. Some people have only a few pain crises a year. Others have a dozen or more a year. A severe pain crisis requires a hospital stay.
Some people with sickle cell anemia also have chronic pain from bone and joint damage, ulcers, and other causes.
- Swelling of hands and feet. Sickle-shaped red blood cells block blood circulation in the hands and feet, which can cause them to swell.
- Frequent infections. The spleen is important for protecting against infections. Sickle cells can damage the spleen, raising the risk of developing infections. Babies and children with sickle cell anemia commonly receive vaccinations and antibiotics to prevent potentially life-threatening infections, such as pneumonia.
- Delayed growth or puberty. Red blood cells provide the body with the oxygen and nutrients needed for growth. A shortage of healthy red blood cells can slow growth in babies and children and delay puberty in teenagers.
- Vision problems. Tiny blood vessels that supply blood to the eyes can become plugged with sickle cells. This can damage the portion of the eye that processes visual images, called the retina, and lead to vision problems.
When to see a doctor
See your healthcare professional right away if you or your child has symptoms of sickle cell anemia, including fever or stroke.
Infections often start with a fever and can be life-threatening. Because children with sickle cell anemia are prone to infections, seek prompt medical attention for a fever greater than 101.5 degrees Fahrenheit (38.5 degrees Celsius).
Seek emergency care for symptoms of stroke, which include:
- One-sided paralysis or weakness in the face, arms or legs.
- Confusion.
- Difficulty walking or talking.
- Sudden vision changes.
- Unexplained numbness.
- Severe headache.
Causes
Sickle cell anemia is caused by a change in the gene that tells the body to make hemoglobin. Hemoglobin is the iron-rich compound in red blood cells that allows these cells to carry oxygen from the lungs to the rest of the body. The hemoglobin associated with sickle cell anemia causes red blood cells to become rigid, sticky and misshapen.
For a child to have sickle cell anemia, both parents must carry one copy of the sickle cell gene and pass both copies to the child.
If only one parent passes the sickle cell gene to the child, that child will have the sickle cell trait. With one typical hemoglobin gene and one sickle cell gene, people with the sickle cell trait make both typical hemoglobin and sickle cell hemoglobin.
Their blood might contain some sickle cells, but they generally don't have symptoms. They're carriers of the disease. That means they can pass the gene to their children.
Risk factors
For a baby to have sickle cell anemia, both parents must carry a sickle cell gene. In the United States, sickle cell anemia most commonly affects people of African, Mediterranean and Middle Eastern descent.
Complications
Sickle cell anemia can lead to a host of complications, including:
- Stroke. Sickle cells can block blood flow to the brain. Signs of stroke include seizures, weakness or numbness of the arms and legs, sudden speech difficulties, and loss of consciousness. If your child has any of these signs or symptoms, seek medical treatment right away. A stroke can be fatal.
- Acute chest syndrome. A lung infection or sickle cells blocking blood vessels in the lungs can cause this life-threatening complication. Symptoms include chest pain, fever and difficulty breathing. Acute chest syndrome might need emergency medical treatment.
- Avascular necrosis. Sickle cells can block the blood vessels that supply blood to the bones. When the bones don't get enough blood, joints may narrow and bones can die. This can happen anywhere but most often happens in the hip.
- Pulmonary hypertension. People with sickle cell anemia can develop high blood pressure in their lungs. This complication usually affects adults. Shortness of breath and fatigue are common symptoms of this condition, which can be fatal.
- Organ damage. Sickle cells that block blood flow to organs deprive the affected organs of blood and oxygen. In sickle cell anemia, blood also is low in oxygen. This lack of oxygen-rich blood can damage nerves and organs, including the kidneys, liver and spleen, and can be fatal.
- Splenic sequestration. Sickle cells can get trapped in the spleen, causing it to enlarge. This may cause abdominal pain on the left side of the body and can be life-threatening. Parents of children with sickle cell anemia can learn how to locate and feel their child's spleen for enlargement.
- Blindness. Sickle cells can block tiny blood vessels that supply blood to the eyes. Over time, this can lead to blindness.
- Leg ulcers. Sickle cell anemia can cause painful open sores on the legs.
- Gallstones. The breakdown of red blood cells produces a substance called bilirubin. A high level of bilirubin in the body can lead to gallstones.
- Priapism. Sickle cell anemia can cause painful, long-lasting erections, known as priapism. Sickle cells can block the blood vessels in the penis, which can lead to impotence over time.
- Deep vein thrombosis. Sickled red blood cells can cause blood clots, increasing the risk of a clot lodging in a deep vein, known as deep vein thrombosis. It also increases the risk of a blood clot lodging in a lung, known as pulmonary embolism. Either can cause serious illness or even death.
- Pregnancy complications. Sickle cell anemia can increase the risk of high blood pressure and blood clots during pregnancy. It also can increase the risk of miscarriage, premature birth and low birth weight babies.
Prevention
If you carry the sickle cell trait, it can help to see a genetic counselor before you get pregnant. A counselor can help you understand your risk of having a child with sickle cell anemia. You also can learn about possible treatments, preventive measures and reproductive options.
Diagnosis
A blood test can check for the form of hemoglobin that underlies sickle cell anemia. In the United States, this blood test is part of routine newborn screening. But older children and adults can get the test too.
In adults, a blood sample is taken from a vein in the arm. In young children and babies, the blood sample is usually collected from a finger or heel. The sample then goes to a laboratory to be screened for the sickle cell form of hemoglobin.
If you or your child has sickle cell anemia, your healthcare professional might suggest other tests to check for possible complications of the disease.
If you or your child carries the sickle cell gene, you'll likely be referred to a genetic counselor.
Assessing stroke risk
A special ultrasound machine can reveal stroke risk in children. The test uses sound waves to measure blood flow to the brain. This painless test can be used in children as young as 2 years old. Regular blood transfusions can decrease stroke risk.
Tests to detect sickle cell genes before birth
Sickle cell disease can be diagnosed in an unborn baby by sampling some of the amniotic fluid surrounding the baby in the womb. If you or your partner has sickle cell anemia or the sickle cell trait, ask your healthcare team about this screening.
Treatment
Management of sickle cell anemia is usually aimed at avoiding pain episodes, relieving symptoms and preventing complications. Treatments might include medicines and blood transfusions. For some children and teenagers, a stem cell transplant might cure the disease. Gene therapies also are being developed that may offer cures for people with sickle cell disease.
Medicines
- Hydroxyurea (Droxia, Hydrea). Daily hydroxyurea reduces the frequency of pain crises and might reduce the need for blood transfusions and hospital stays. But it can increase the risk of infections. Don't take the drug if you're pregnant.
- L-glutamine oral powder (Endari). It helps in reducing the frequency of pain crises.
- Crizanlizumab (Adakveo). This medicine, given by injection, can help reduce the frequency of pain crises in adults and in children older than 16 years. Side effects can include nausea, joint pain, back pain and fever.
- Voxelotor (Oxbryta). This medicine is used to treat sickle cell disease in adults and in children older than 12 years. Taken by mouth, this medicine can lower the risk of anemia and improve blood flow throughout the body. Side effects can include headache, nausea, diarrhea, fatigue, rash and fever.
- Pain-relieving medicines. Your healthcare professional might prescribe narcotics to help relieve pain during sickle cell pain crises.
Preventing infections
Children with sickle cell anemia might receive penicillin from about 2 months old to 5 years old, or longer. This medicine can help prevent infections, such as pneumonia, which can be life-threatening to children with sickle cell anemia.
Adults who have sickle cell anemia might need to take penicillin throughout their lives if they've had pneumonia or surgery to remove the spleen.
Childhood vaccinations are important for preventing disease in all children. Vaccinations are even more important for children with sickle cell anemia because their infections can be severe.
Your child's healthcare team should make sure that your child gets all the recommended childhood vaccinations. These include vaccines against pneumonia, meningitis, hepatitis B and a yearly flu shot. Vaccines also are important for adults with sickle cell anemia.
During global health threats, such as the COVID-19 pandemic, people with sickle cell anemia should take extra precautions. These include staying at home as much as possible and for those who are eligible, getting vaccinated.
Surgical and other procedures
-
Blood transfusions. Red blood cell transfusions are used to treat and prevent complications, such as stroke, in people with sickle cell disease.
In this procedure red blood cells are removed from a supply of donated blood, then given through a vein to a person with sickle cell anemia. This increases the number of red blood cells that are not affected by sickle cell anemia. This helps reduce symptoms and complications.
Risks include an immune response to the donor blood, which can make it hard to find future donors. Infection and excess iron buildup in the body are other risks. Because excess iron can damage your heart, liver and other organs, you might need treatment to reduce iron levels if you undergo regular transfusions.
-
Stem cell transplant. This also is known as a bone marrow transplant. The procedure involves replacing bone marrow affected by sickle cell anemia with bone marrow from a donor. The procedure usually uses a matched donor, such as a sibling, who doesn't have sickle cell anemia.
A stem cell transplant can cure sickle cell anemia. Stem cell transplant is recommended only for people, usually children, who have significant symptoms and complications of sickle cell anemia. The risks associated with the procedure are high and include death.
-
Stem cell gene addition therapy. In this treatment option, the person's own stem cells are removed, and a gene to produce typical hemoglobin is injected. The stem cells are then given back to the person in a process known as autologous transplant. This option may be a cure for people with sickle cell disease who do not have a well-matched donor.
-
Gene editing therapy. This Food and Drug Administration (FDA)-approved treatment works by making changes to the DNA in a person's stem cells. Stem cells are removed from the body, and the sickle gene is changed, also called edited, to help restore the cells' ability to make healthy red blood cells. The treated stem cells are then returned to the body through the blood. This is called an infusion.
People who are successfully treated with gene editing therapy no longer have symptoms of sickle cell disease. This treatment is FDA-approved for people 12 years old and older. Long-term effects of this new treatment are not yet known and will continue to be studied.
Clinical trials are ongoing to address stem cell transplantation in adults and gene therapies.
Self care
The following steps to stay healthy might help you avoid complications of sickle cell anemia:
- Take folic acid supplements daily and choose a healthy diet. Bone marrow needs folic acid and other vitamins to make new red blood cells. Ask your healthcare team about a folic acid supplement and other vitamins. Eat a variety of colorful fruits and vegetables, as well as whole grains.
- Drink plenty of water. Dehydration can increase your risk of a sickle cell pain crisis. Drink water throughout the day, aiming for about eight glasses a day. Increase the amount of water that you drink if you exercise or spend time in a hot, dry climate.
- Avoid temperature extremes. Exposure to extreme heat or cold can increase your risk of a sickle cell pain crisis.
- Exercise regularly, but don't overdo it. Ask your healthcare professional how much exercise is right for you.
- Use medicine with caution. Use pain medicines such as ibuprofen (Advil, Motrin, others) or naproxen sodium (Aleve) sparingly, if at all, because of the possible effect on the kidneys. Ask your healthcare professional before taking any medicine you can get without a prescription.
- Don't smoke. Smoking increases your risk of pain crises.
Coping and support
If you or someone in your family has sickle cell anemia, these ideas might help you cope:
- Finding someone to talk with. Living with a chronic illness is stressful. Consider consulting a mental health professional, such as a psychologist, counselor or social worker, to help you cope.
- Join a support group. Ask your healthcare team about support groups for families in your area. Talking with others who face challenges similar to yours can be helpful.
- Explore ways to cope with the pain. Pain medicine can't always take away all the pain. Work with your healthcare professional to find ways to control your pain. You might try heating pads, hot baths, massages or physical therapy.
- Learning about sickle cell anemia to make informed decisions about care. Learn as much as you can about the disease. Ask questions during your child's appointments. Ask your healthcare team to recommend good sources of information.
Preparing for your appointment
Sickle cell anemia is usually diagnosed through genetic screening done when a baby is born. Those test results will likely be given to your primary healthcare professional, who may refer you to a doctor who specializes in blood disorders, called a hematologist, or a pediatric hematologist.
Here's information to help you get ready for your appointment.
What you can do
Make a list of:
- Your symptoms, including any that seem unrelated to the reason for which you scheduled the appointment, and when they began.
- Key personal information, including family medical history and whether anyone has sickle cell anemia or has the trait for it.
- Questions to ask your healthcare team.
Bring a family member or friend along, if possible, to help you remember the information you're given.
For sickle cell anemia, questions to ask your healthcare professional include:
- What's the most likely cause of the symptoms?
- Are there other possible causes?
- What tests are needed?
- What treatments are available, and which do you recommend?
- What side effects are common with these treatments?
- Are there other treatment options available?
- How likely is the treatment to work?
- Are there and food or activity limitations?
- Do you have brochures or other printed material that I can have? What websites do you recommend?
Don't hesitate to ask other questions.
What to expect from your doctor
Your healthcare professional is likely to ask you questions, including:
- When did you notice symptoms?
- Have they been continuous or occasional?
- What, if anything, seems to improve symptoms?
- What, if anything, seems to worsen them?

